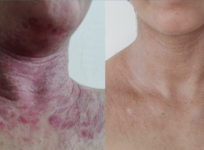


The first wave of data from FDA's new Adverse Events Reporting System for dietary supplements shows two important things: The system works, enabling the FDA to act quickly when there's evidence of life-threatening side effects; and serious adverse events are rare given how widely supplements are used.


